Research
Pluripotent, Naive, and Blastomere-Like Stem Cell Biology and Reprogramming
Example Publications:
- Zimmerlin L., et al. (2024). “Proteogenomic reprogramming to a functional human totipotent stem cell state via a PARP-DUX4 regulatory axis.” bioRxiv.
- Zimmerlin L., et al. (2020). “Pleiotropic roles of tankyrase/PARP proteins in the establishment and maintenance of human naive pluripotency.” Experimental Cell Research.
- Park T.S., et al. (2016). “Tankyrase inhibition promotes a stable human naïve pluripotent state with improved functionality.” Development.
- Huo J.S., et al. (2014). “Cancer-like epigenetic derangements of human pluripotent stem cells and their impact on applications in regeneration and repair.” Current Opinion in Genetics & Development.
- Shah S.N., et al. (2012). “HMGA1 reprograms somatic cells into pluripotent stem cells by inducing stem cell transcriptional networks.” PLoS One.
After hiPSC became available in 2007, the Zambidis lab has developed a highly cited and commercialized platform (Life Technologies, ThermoFisher) for efficiently generating nonintegrated, human induced pluripotent stem cell (hiPSC) lines from CD33+ human myeloid progenitors (Burridge et al, 2011, Park et al, 2012). We later refined this technology to generate a host of xeno-/feeder-/transgene-free human induced pluripotent stem cell lines from healthy peripheral and cord blood donation, as well as from the cord blood or skin biopsies of patients with various inherited or acquired disease backgrounds (e.g., Park et al, 2017). We have used these lines to study genomic, epigenetic, and functional differences between healthy and diseased iPSC-derived effector cells, such as vascular progenitors, to ultimately offer cGMP-ready therapeutic cells for regenerative medicine applications. A range of these lines has been extensively quality controlled and made available to the community at a low passage number via WiCell. By overcoming lineage priming without aberrant erasure of epigenetic imprints, our novel tankyrase/PARP inhibitor-regulated naïve (TIRN) stem cell system (patent pending) has overcome the lineage bias characteristic of primed pluripotency. This may enable the production of HLA-matched cell therapies (e.g., retinal progenitors, vascular progenitors, immune cell therapies) from a bank of 10-100 highly selected haplotype donors instead of autologous production (Taylor et al., 2005). The development of such TIRN stem cell haplobanks may finally ‘keep the promise’ of iPSC-derived individualized therapies while avoiding the exuberant cost of patient-specific cGMP-grade iPSC reprogramming. We are currently expanding our pool of disease-type and healthy clinical grade iPSC and recently also ESC with full consent for future commercialization, using a xeno-/ feeder-/ and transgene-free episomal reprogramming method amenable to cGMP-production (cGMP-ready; manuscript in preparation).
The derivation and maintenance of mammalian pluripotent stem cells in stable naïve pluripotent states has a wide impact in human developmental biology and may have a wide impact in regenerative medicine. However, human PSCs are unstable in classical naïve mouse embryonic stem cell (ESC) WNT and MEK/ERK signal inhibition (2i) culture. We have shown that a broad repertoire of conventional hESC and transgene-independent human induced pluripotent stem cell (hiPSC) lines could be reverted to stable human preimplantation inner cell mass (ICM)-like naïve states with only WNT, MEK/ERK, and tankyrase inhibition (LIF-3i). LIF-3i-reverted hPSCs retained normal karyotypes and genomic imprints, and attained defining mouse ESC-like functional features, including high clonal self-renewal, independence from MEK/ERK signaling, dependence on JAK/STAT3 and BMP4 signaling, and naïve-specific transcriptional and epigenetic configurations. Importantly, L3i TIRN-SC maintained normal karyotypes and imprinted CpG patterns, and were devoid of irreversible demethylation defects (Thomas et al., 2021; Zimmerlin et al., 2016) that were reported in other naïve reversion systems (Pastor et al., 2016; Theunissen et al., 2016), and suggested to be secondary to prolonged MEK inhibition (Choi et al., 2017) as well as HDAC inhibition. Tankyrase inhibition promoted a stable acquisition of a human preimplantation ICM-like ground state via modulation of WNT signaling, and was most efficacious in efficiently reprogrammed conventional hiPSCs. Importantly, reversion to TIRN-SC eliminated interline variability and significantly enhanced the multi-lineage differentiation performance of isogenic primed hPSC differentiation; without requirement for transitioning back to primed culture conditions to restore differentiation potency (Lee et al., 2017; Sahakyan et al., 2017; Warrier et al., 2017). On the contrary, we have observed direct differentiation of TIRN stem cells (TIRN-SC) towards vascular, hematopoietic, and retinal lineages in 2D and 3D protocols, as well as in non-directed teratoma assays, with greatly improved differentiation efficiency and reduced lineage bias; all but abrogating the phenomenon of ‘non-permissive lines’ for certain differentiation pathways (Zhong et al, 2014, Zambidis et al, 2020, Zimmerlin et al, 2022). The TIRN stem cell system has become the backbone of our scientific endeavor and we expect this stem cell system to have great utility in regenerative medicine and human disease modeling.
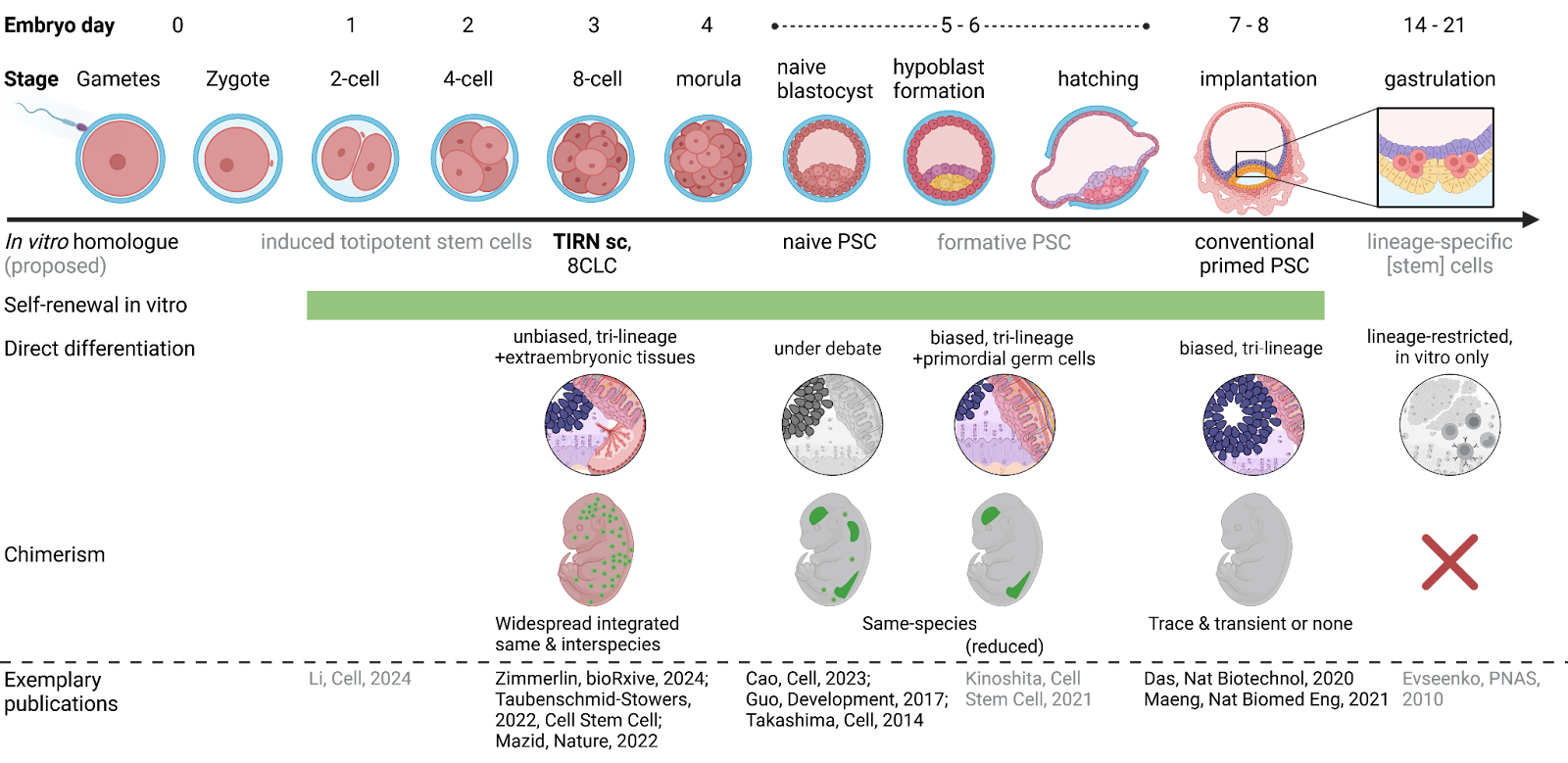
When interrogating the developmental status of our original L3i and improved feeder- & xeno-free TIRN stem cells, we discovered a hybrid transcriptional program of stable naive epiblast-like pluripotency together with features of four-to-eight cell blastomere-like totipotency (also see Proteogenomic regulation of human stem cells with a focus on the PARP-Dux4 regulatory axis). Other than conventional hiPSC, human L3i and improved L4i (manuscript in preparation; patent pending) TIRN cells integrated into mouse embryos with up to 15% chimerism in all tissues on embryonic day 14.5 (Zimmerlin et al, 2024 [bioRxiv]; now accepted at Cell Reports, stay tuned!). We have successfully outgrown human hematopoietic and neural cells from chimeric embryos. To avoid the ethical challenges especially of neural and germline interspecies chimerism, we have decided to not let embryos develop through birth but instead commenced studies to lineage-restrict the human cell contribution while also preserving the excellent chimeric integration and persistence of TIRN-SC (see Stem Cell-based Hematopoietic, Vascular, and Retinal Regeneration).
Proteogenomic regulation of human stem cells with a focus on the PARP-Dux4 regulatory axis
Example Publications:
- Zimmerlin L., et al. (2024). “Proteogenomic reprogramming to a functional human totipotent stem cell state via a PARP-DUX4 regulatory axis.” bioRxiv.
- Huo J.S., et al. (2014). “Cancer-like epigenetic derangements of human pluripotent stem cells and their impact on applications in regeneration and repair.” Current Opinion in Genetics & Development.
PARP1 and TNKS1/2 are PARP family enzymes that catalyze post-translational poly-ADP-ribose (PAR)ylation of protein substrates and play formative roles in regulating chromatin structure, transcription, and ubiquitination in not only adult cells, but also during embryonic development (Zimmerlin and Zambidis, 2020). Chemical inhibition of PARylation impairs the homeostasis and progression of preimplantation embryonic development (Imamura et al., 2004); failure to degrade PAR is embryonic lethal (Koh et al., 2004). TNKS1/2 activity controls developmental progression of zygotic genome activation (ZGA) (Gambini et al., 2020), and PARP1 regulates histone ubiquitination, DNA methylation, RNA processing, DNA repair during ZGA (Hamazaki et al., 2015; Osada et al., 2013; Tsai et al., 2023), cellular reprogramming (Chiou et al., 2013; Doege et al., 2012; Jiang et al., 2015), pluripotency (Roper et al., 2014), trophoblast specification (Hemberger et al., 2003) and chromatin plasticity (Ciccarone et al., 2017; Ko and Ren, 2012) during preimplantation embryogenesis. Although loss of a single PARP protein minimally impairs development (suggesting redundancy of PARP proteins), compound TNKS1/TNKS2 and PARP1/PARP2 knockouts, or dominant-negative PARP1 are embryonic lethal (Chiang et al., 2008; Hsiao et al., 2006; Menissier de Murcia et al., 2003; Shao et al., 2023)
PARP1 and TNKS1/2 both possess catalytic and non-catalytic activities via multifunctional protein domains. TNKS1/2 proteins similarly mediate non-PAR-catalytic activities such as DNA repair regulation and pexophagy/autophagy (Bhardwaj et al., 2017; Li et al., 2017; Nagy et al., 2016). Furthermore, the catalytic activities of TNKS and PARP1 deeply impact homeostasis of the proteome (proteostasis) during embryogenesis, via extensive networks of post-translational PAR-dependent ubiquitination (PARdU) (Callow et al., 2011; Cho-Park and Steller, 2013; Rona et al., 2018; Vittal et al., 2015; Vivelo et al., 2019). Beyond the UPS, post-translational, UPS-independent (Mukhopadhyay and Riezman, 2007; Trulsson et al., 2022) ubiquitination structurally regulates chromatin/histone architecture (Vaughan et al., 2021), and transcription factor DNA binding (Mark and Rape, 2021) including of the core pluripotency factors NANOG (Kim et al., 2014; Pei, 2017), OCT4 (Baek et al., 2020; Li et al., 2018; Rhie et al., 2021) and SOX2 (Cui et al., 2018; Fang et al., 2014a; Wang et al., 2016).
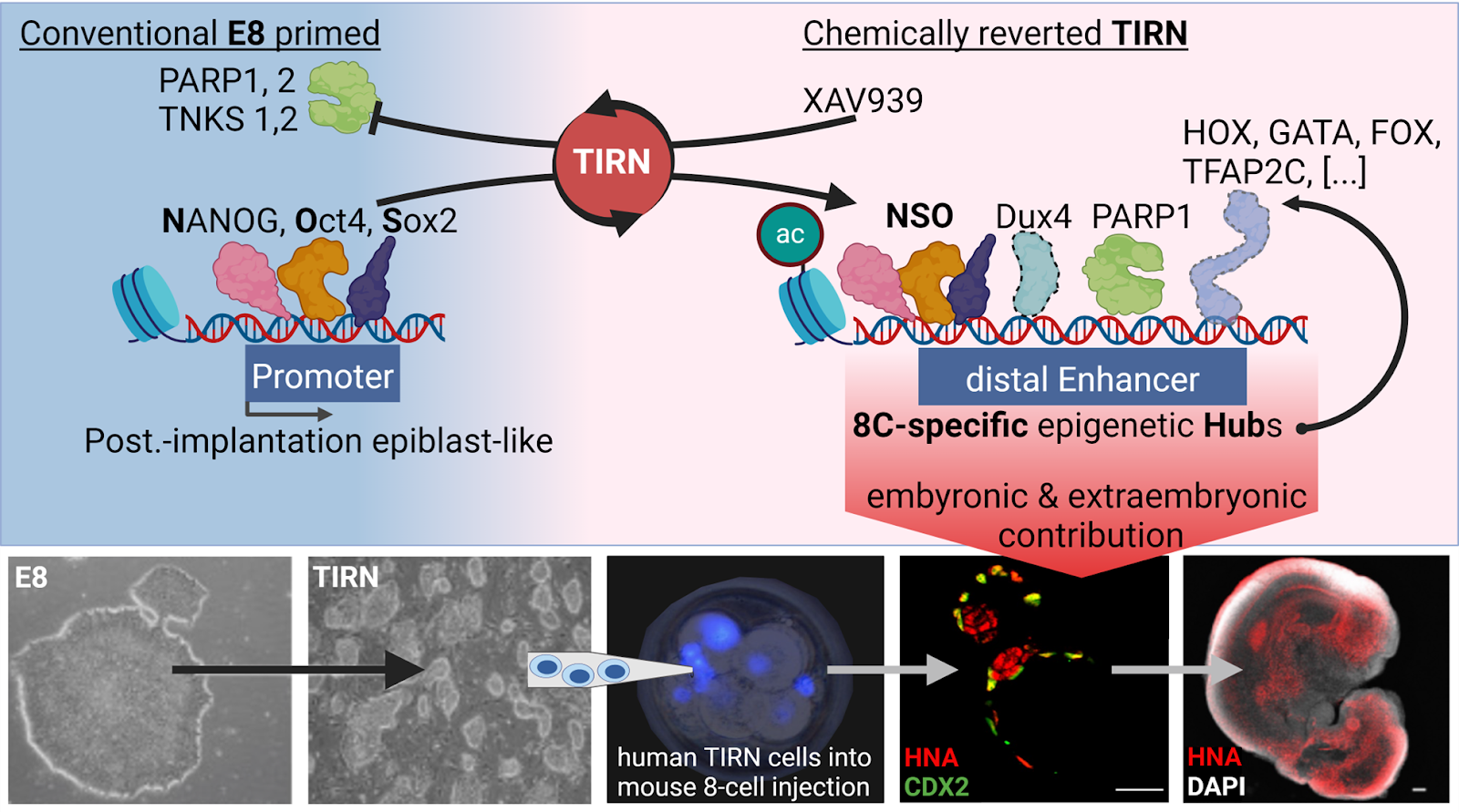
Several studies have employed chemical PARP inhibition to expand pluripotency and improve the chimeric contribution of mouse embryonic stem cells (mESC) into embryos (Gao et al., 2019; Liu et al., 2021; Yang et al., 2017a; Yang et al., 2017b), or to improve functional pluripotency (Park et al., 2020; Park et al., 2018; Thomas et al., 2021; Zimmerlin et al., 2016; Zimmerlin et al., 2017). We have first demonstrated (Zimmerlin et al., 2016) that primed, conventional human pluripotent stem cells (hPSC) can be chemically reverted to a naive epiblast-like pluripotent state with improved differentiation potential via a two-step chemical reprogramming system (‘LIF-5i -> LIF-3i’), that comprised of sequential culture with LIF and five small molecules (i.e., Hedgehog signalling activation, cAMP agonism, GSK3ϕ3 inhibition, MEK inhibition, and nonspecific PARP inhibition (XAV939)). XAV939 is a promiscuous small molecule that inhibits in vitro PARylation activities of not only TNKS1 and TNKS2 (IC50 ∼5.2-94.6 nM), but also PARP1 and PARP2 (IC50 ∼26.9-169 nM) (Huang et al., 2009; Thorsell et al., 2017). Sequential naïve reversion with continuous exposure to XAV939 reprogrammed a large repertoire of >25 conventional lineage-primed hPSC to the TIRN stem cell state (Park et al., 2020; Park et al., 2018; Thomas et al., 2021; Zimmerlin et al., 2016; Zimmerlin et al., 2017, also see Pluripotent, Naive, and Blastomere-Like Stem Cell Biology and Reprogramming). TIRN-SC possess high clonal proliferation, phosphorylated-STAT3 signalling, MEK-ERK/bFGF signalling independence, modulation of ϕ3-catenin expression, and erasure of lineage-primed gene expression (Park et al., 2020; Zimmerlin et al., 2016). The cells were globally inhibited in both TNKS and PARP1 PARylation activities, and TNKS1/2 protein levels were increased while PARP1 protein levels decreased; potentially defining a novel functional PARP protein equilibrium (Zimmerlin et al, 2024 [bioRxiv]). TIRN-SC underwent a global ubiquitinome reprogramming that was driven by hundreds of PARP1 and TNKS1/2 targets; including core factors NANOG and SOX2 and epigenetic modifiers. Unexpectedly, XAV939-mediated TIRN reprogramming activated simultaneous, single-cell co-expressions of ZGA-priming (e.g., TPRXL) and DUX4 gene targets, as well as a panoply of PARP1-regulated lineage-specifying transcriptional factors.
The cells also underwent a genome-wide epigenetic reprogramming driven by reduction and redistribution of PARP1 chromatin co-binding to core pluripotency factors NANOG/SOX2/OCT4 (NSO) at gained promoter, intragenic, and distal enhancer sites of 8C- and morula-associated genes (e.g., EBF2, HOXB2, GATA6). We identified a cohort of gained NSO and PARP1 (NSOP) co-binding enhancer sites near transcriptionally active, as well as distal regions of DUX4-accessible chromatin. Remarkably, these gained NSOP-DUX4 co-binding sites were further enriched in co-binding motifs for hundreds of the same embryonic and extraembryonic lineage-determining pioneer factors that were actively and concurrently already co-expressed in TIRN-SC, suggesting the discovery of candidate totipotency-regulating chromatin regions.
Translational Stem Cell-based Hematopoietic, Vascular, and Retinal Regeneration
Example Publications:
- Park T.S., et al. (2014). “Vascular progenitors from cord blood–derived induced pluripotent stem cells possess augmented capacity for regenerating ischemic retinal vasculature.” Circulation.
- Zhong X., et al. (2014). “Generation of three-dimensional retinal tissue with functional photoreceptors from human iPSCs.” Nature Communications.
- Zambidis E.T., et al. (2008). “Expression of angiotensin-converting enzyme (CD143) identifies and regulates primitive hemangioblasts derived from human pluripotent stem cells.” Blood.
- Zimmerlin L., et al. (2022). “Improved generation and long-term engraftment of retinal organoids from Tankyrase/PARP-Inhibitor-Regulated Naïve human pluripotent stem cells (TIRN-hPSC).” Investigative Ophthalmology & Visual Science.
As a fellow, I was one of the first investigators at Johns Hopkins to work with human embryonic stem cells (hESC) after the federal government made them available to the biomedical community in 2002. My long term goal is to understand the developmental and molecular mechanisms that regulate human pluripotent and totipotent-like stem cell (hPSC) differentiation to clinically important lineages, including vascular, hematopoietic, retinal, neural, and cardiac tissues for regenerative medicine applications.
In 2004, I received an NIH K08 clinician scientist award, specifically to develop a model system of human hemato-vascular development based human pluripotent stem cells. In 2005, I was the first to identify and characterize the phenotype and functionality of primitive human hemangioblast generated in vitro from differentiating hESC (Zambidis et al, 2005, 2008). This novel experimental system models the events of early human yolk sack type hemo-vasculogenesis. Based on this, my lab has developed hiPSC-based vascular therapies for treating retinopathies (Park et al, 2014), which we later refined for improved functional engraftment and persistence (Park et al, 2017) using our TIRN stem cell system.
Our work on vascular progenitors and derived therapies particularly focuses on regenerating the microvascular damage of diabetic retinopathy. We have reprogrammed multiple iPSC-lines from diabetes patients (also see Pluripotent, Naive, and Blastomere-Like Stem Cell Biology and Reprogramming) and in 2014 complemented this work by generating retinal vasculature and photoreceptors from the same diabetic hiPSC-lines (Park et al, 2014).
The TIRN system was the key to overcome the lineage bias of non-permissive iPSC-lines, enabling the reliable generation of retinal organoids from all tested stem cell lines (Zhong et al, 2014 & Zimmerlin et al, 2016). While testing the functional integration of human and mouse retina at embryonic, neonatal, and adult stages, we have recently isolated a highly prolific committed retinal progenitor cell, which can chimerize mouse embryos and generate all cell types of the neuroretina for weeks (manuscript in preparation). When incorporated into eye-ablation mutant animals via blastocyst complementation, this technology may allow the generation of a fully structured human retina without the ethical challenge of off-target integration in interspecies chimeras. We have also defined a putative mes-endoderm stem cell generating lineage restricted teratoma without ectoderm structures and demonstrated their preserved interspecies chimerization after mouse blastocyst injection. This strategy could eventually provide fully human mes-endoderm derived transplant organs including heart, liver, pancreas, lung, and possibly kidneys, as well as generate neigh unlimited amounts of blood and cell products (also see Immunotherapy incl. Stem Cell-based CAR therapies) in livestock animals while circumventing the ethical challenges associated with neural or germline integration.
Immunotherapy incl. Stem Cell-based CAR therapies
Example Publications:
- Buys W., Zambidis E.T. (2023). “Harnessing bioengineered myeloid progenitors for precision immunotherapies.” npj Regenerative Medicine.
- Cui C., et al. (2020). “Adoptive transfer of immature myeloid cells lacking NF-κB p50 (p50-IMC) slows high-risk neuroblastoma tumor growth.” Journal for ImmunoTherapy of Cancer.
Granulocytes and macrophages are the frontline defenders of the innate immune system. These myeloid cells play a crucial role in not only eliminating pathogens and tumor cells, but also regulating adaptive immune responses. In neonatal sepsis and post-chemotherapy agranulocytosis, the absence of these cells leaves the host highly vulnerable to infections. Beyond replacement to prevent or control neutropenic sepsis, engineered myeloid cells may offer distinct opportunities for cell therapies. For example, the mobility and specific homing capacities of neutrophils to sites of inflammation could be exploited to deliver biocidal agents, or anti-inflammatory healing signals during sepsis, autoimmunity, and organ transplantation. Additionally, myeloid cells can be engineered to express chimeric antigen receptors (CAR), carry chemotherapeutics, or enhance lymphoid tumor killing. However, donation-derived granulocytes and macrophages cannot be produced in sufficient cell numbers for many of these applications, and their short lifespan further complicates logistics and efficacy. In 2023, we have proposed to instead use multipotent granulocyte macrophage progenitors manufactured from hiPSC (iGMP) or from primary CD34+. Such iGMP may not only alleviate the sourcing difficulties of short lived myeloid effector cells, but also extend the duration of effect, greatly simplify cell engineering, and allow an ‘off-the-shelf’ provision, due to superior in vivo expansion capacities and improved cryo-preservation tolerance. By reducing the required cell numbers, bio-engineered, short-term engrafting myeloid progenitors could help bring bioreactor volumes down to a practical scale for making their production more affordable (Buys, Zambidis 2023). We propose that these collective advantages ultimately outweigh the higher initial investment for establishing engineered hiPSC lines or harvesting donor primary CD34+ hematopoietic stem and progenitor cells; particularly when combined with haplotype stem cell banking strategies (see Pluripotent, Naive, and Blastomere-Like Stem Cell Biology and Reprogramming).
We have already developed a protocol generating iGMP at a 5-to-50-fold greater efficiency per cell culture volume and per input cell from TIRN-SC than from isogeneic parallel conventional E8 cultures (manuscript in preparation). TIRN-SC derived iGMP produced approx. 10-fold more granulocyte-macrophage colonies (CFU-GM) in methylcellulose assays, than parallel conventional E8 progeny. Derived effector cells demonstrated similar phagocytosis and LPS stimulated TNF production and greatly improved chemotaxis and respiratory burst, the latter two even surpassing primary adult monocytes (manuscript in preparation). We have now begun functionalizing our cord blood derived cGMP-ready iPSC-lines with chimeric antigen receptors against various solid tumor antigens (rev. Buys, Zambidis 2024) and against non-physiological adaptor molecules that may enable facile and quick re-targeting of derived effector cells against a plethora of tumor targets, including GD2, EGFR, and Her2.
Clinical Application of Hematopoietic Stem Cell Transplantation in Malignant and Non-Malignant Contexts
Example Publications:
- Klein O.R., et al. (2021). “Reduced intensity bone marrow transplantation with post-transplant cyclophosphamide for pediatric inherited immune deficiencies and bone marrow failure syndromes.” Journal of Clinical Immunology.
- Symons H.J., et al. (2020). “Myeloablative haploidentical BMT with posttransplant cyclophosphamide for hematologic malignancies in children and adults.” Blood Advances.
Adjacent to my clinical work in pediatric bone-marrow transplantation at the Johns Hopkins Sidney Kimmel Comprehensive Cancer Center, I have contributed to advancing hematopoietic cell transplantation (HCT) through the clinical implementation of post-transplant cyclophosphamide (PTCy), refining protocols for both pediatric and adult patients. My work helped demonstrate improved safety and efficacy in haploidentical bone marrow transplants, including reduced-intensity approaches (Klein et al., 2021), offering a feasible treatment option for individuals lacking a beneficial HLA-match (Symons et al., 2020).
Above all, the mission of my lab at “America’s first research University”, is bridging the translational gap between basic health science and applied medicine. The generosity and excitement of my patients to contribute to the research mission have enabled me to build a sizeable stem cell bank of various inherited disease backgrounds, to elucidate mechanisms of lysosomal dysfunction and neurodevelopmental abnormalities (Srikanth et al., 2021; Awad et al., 2017; also see Pluripotent, Naive, and Blastomere-Like Stem Cell Biology and Reprogramming); a work which is still ongoing using our updated episomal reprogramming method (manuscript in preparation).